Workflow(1) Preprocess
Workflow(1) Preprocess
本篇为蛋白组数据处理第一篇,整体讲一下流程
This is the first chapter of the proteomics data analysis process.
准备工作:
1.下载Perseus(download Perseus)
具体步骤:(Steps)
1.peaks搜库(database searching)
2.打开Perseus(turn to Perseus)
3.点击上传数据(Upload your data)
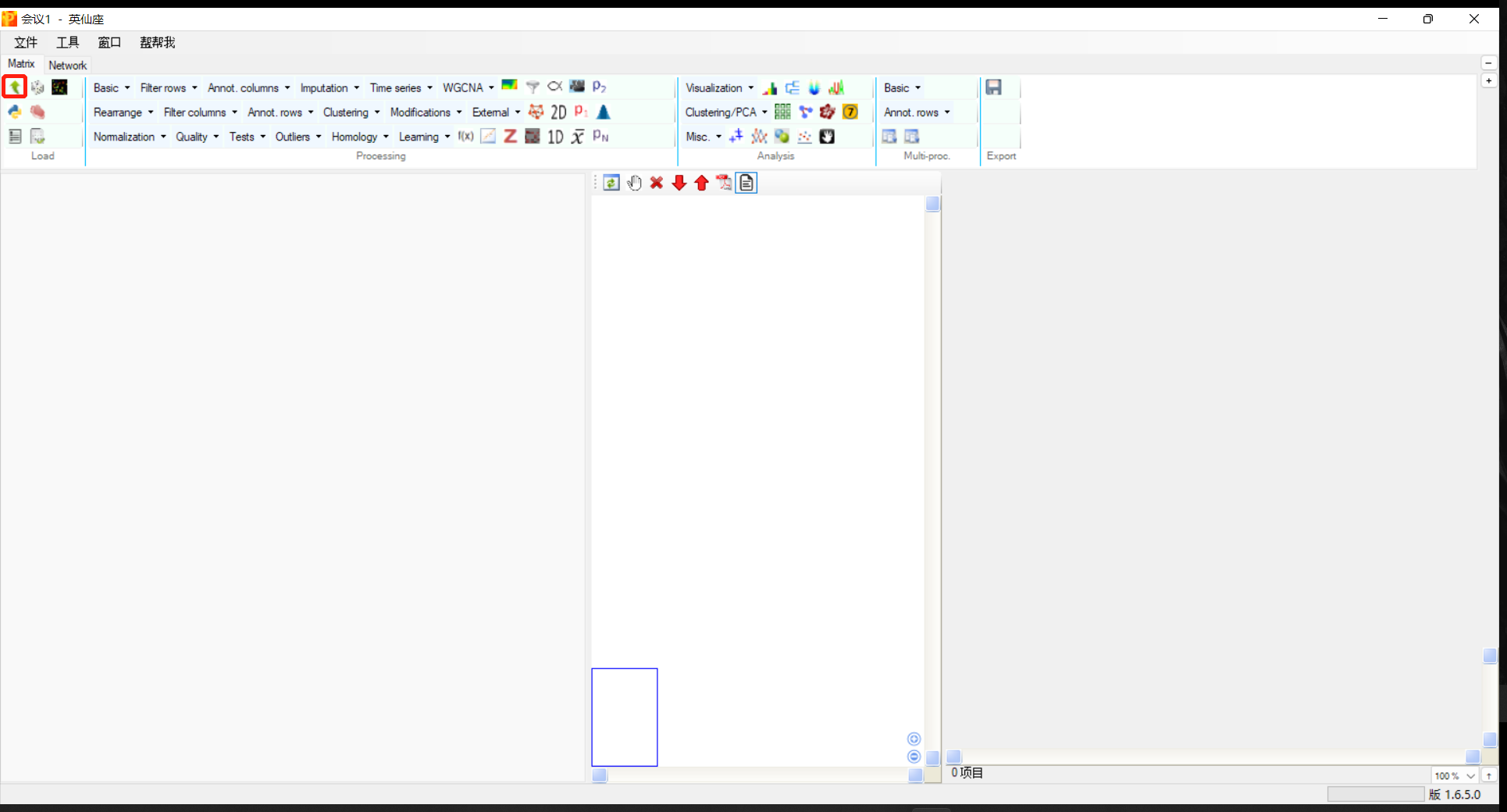
4.peaks搜库结果文件为csv,更改右下角数据格式以发现文件(choose the right format of your data in the window)
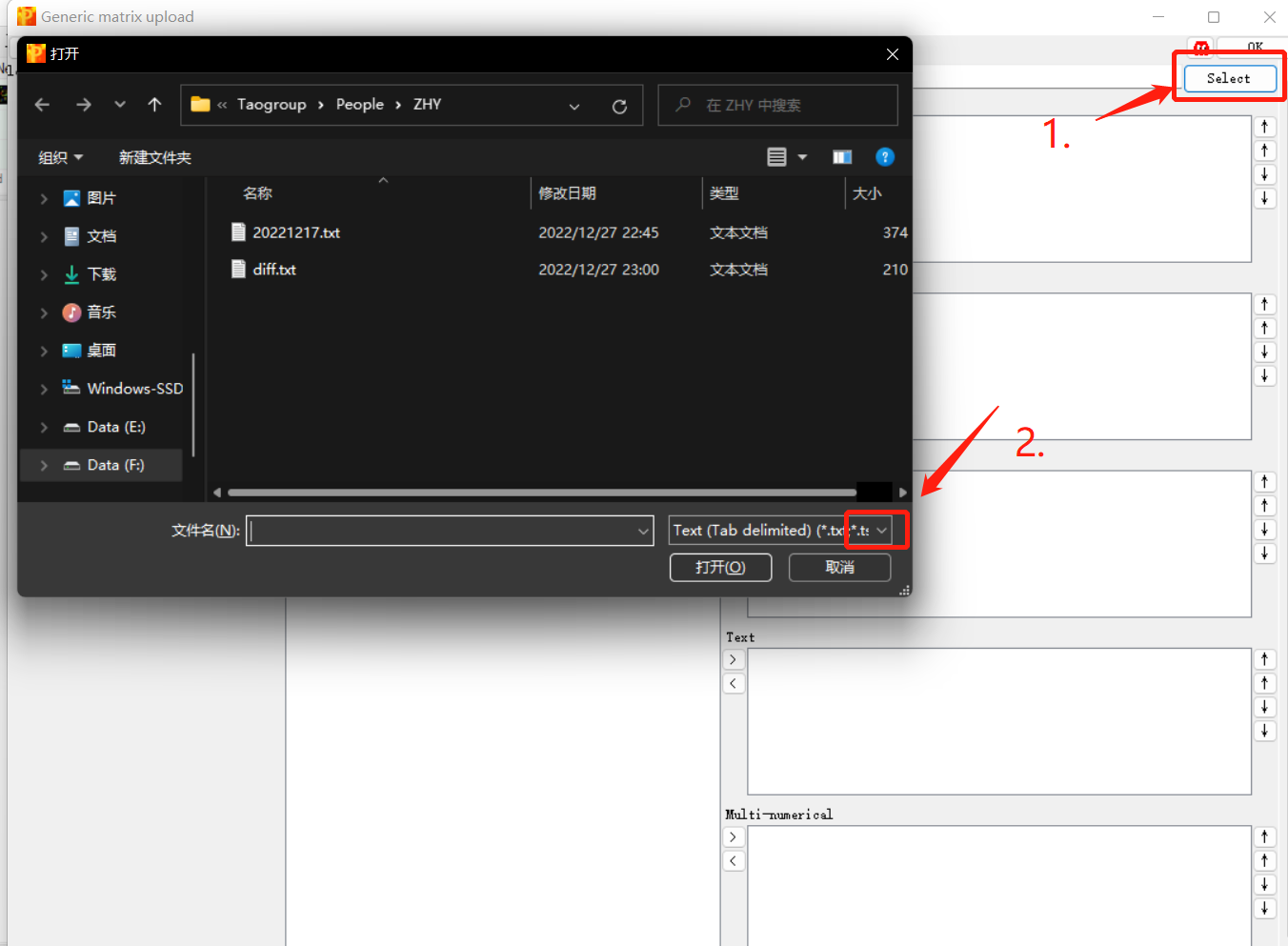
5.导入数据(input your data)
6.对数据进行log转换(do the log transformation)
Basic->transform->ok
**注意(Attention)**:这里默认的就是log2,如果有可靠依据,需要其他log可自行更改式
The default log transformation is log2, if there is any solid reason, you can change by your need.
7.过滤无效蛋白(filter invalid value protein)
Filter rows -> Filter rows based on valid values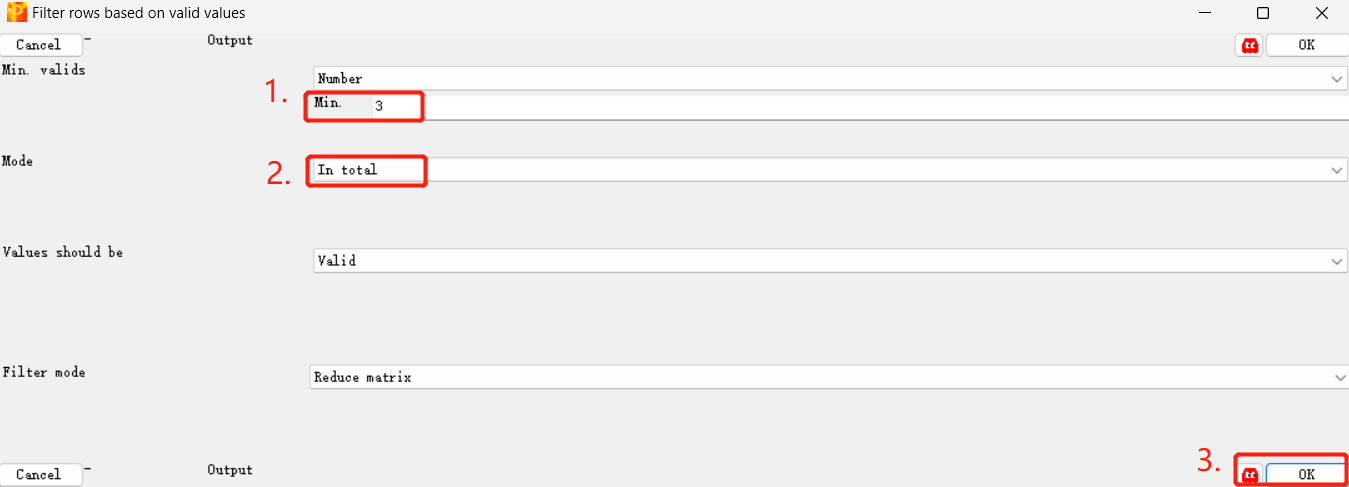
7.1.处数值最少为样本总数的一半,如果留下的蛋白较多,可以适当提高标准提高最少数量。(The valued number of should be at least half of the samples)
7.2.Mode could be In total or In each group。
注意: 1处和2处的选项要匹配,例如一共有两个组,每组5个样本,共10个样本。如果2处选In total, 1处则填5或5以上的数值;如果2处填In each group, 则1处填3或以上的数值。
8.过滤PTM(filter the PTM)
(如果做全蛋白,则跳过此步骤)(If you did the intact protein analysis, then skip this step)
Filter rows -> filter rows based on categorical column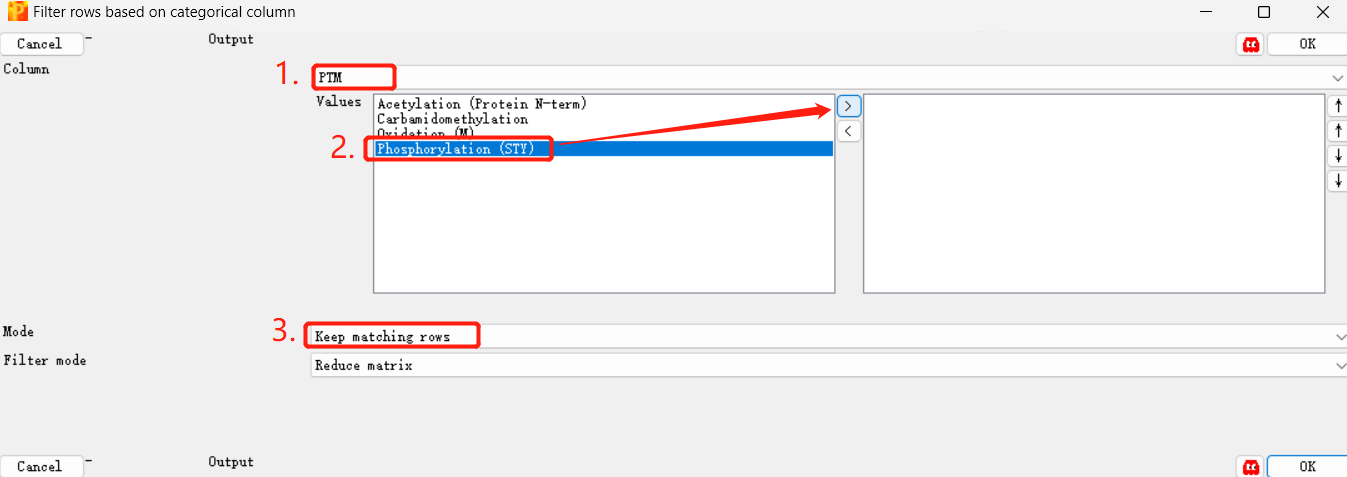
8.1 1处选为PTM,2处根据数据类型选择ptm加入右边的边框,3处选Keep matching rows
9.填补缺失值(impute missing values to make analysis possible)
此步骤填补缺失值与前面筛选有效值有关,如果筛选得严格,则填补缺失值影响不大,如果筛选的宽松,则填补缺失值会对以后差异分析有影响.
一般方法:Imputation -> replace missing values from normal distribution
其余两种也可选择,根据具体需要,by constant则填补固定值,by NaN则填补为NaN,没有大用,并且可能会对生信分析数据识别产生错误。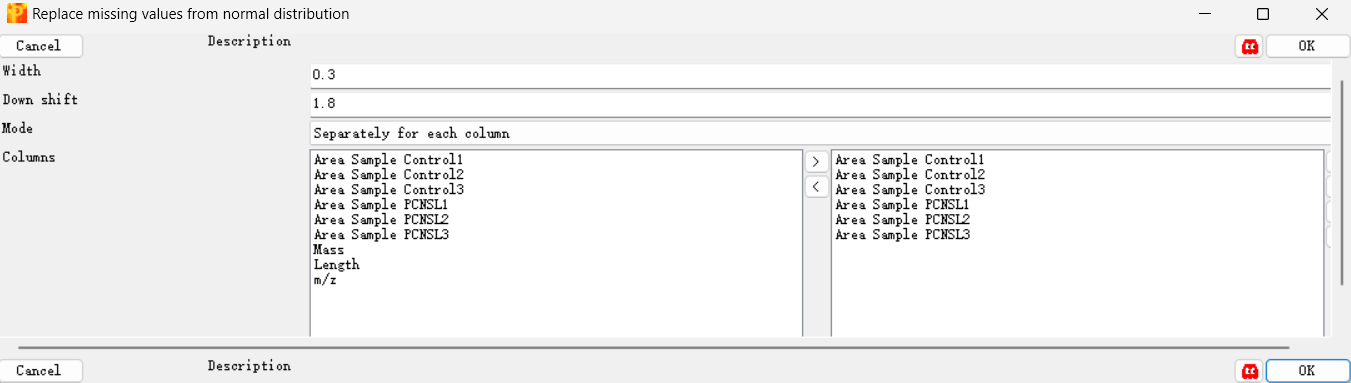
可以看到默认Mode选项是对每列分别计算正态分布,然后填补。
10.归一化(Nomalization)
对列进行归一化,消除样本总量差异带来的intensity的差异
Normalization -> Subtract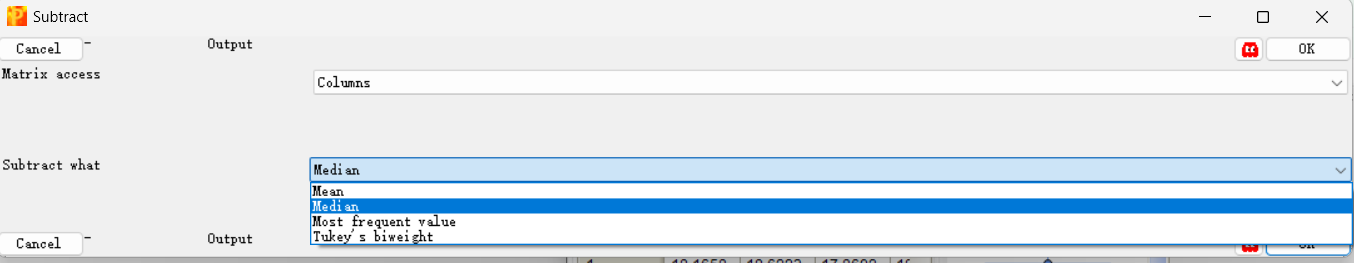
Matrix access选为Colunms
Subtract what选为Median,Mean都可,一般为mean,为除以每列总和除以总蛋白数,因为每组蛋白数是一样的,则是用每组的总数来做归一化,消除总量的影响。
11.导出预处理数值
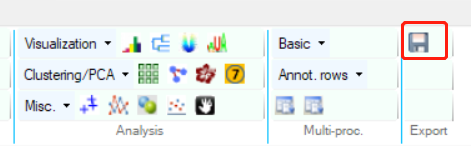
保存处理好的数据最后一步,同时,中间的每一步结果也可以根据需要导出。
注意文件名设为英文
12.打开处理流程文件
同时注意,处理流程也要保留,以供之后查阅。
下次再次打开本次处理流程也从文件处点击打开,步骤为:先打开Perseus,再点击 文件 -> open